ML Models¶
This section summarizes the machine learning approach used in PolyDrug for solubility parameter prediction.
ML Workflow¶
The machine learning workflow follows the approach described in our published study.[1] A combined dataset of small molecules and polymers was prepared, and each entry was represented using molecular, quantum-chemical, and chain-related descriptors. For polymers, repeating-element representations were used so that polymer structures could be encoded in a form suitable for descriptor generation.[2]
The overall workflow includes dataset preparation, descriptor calculation, feature selection, model training, and model evaluation. Feature engineering showed that molecular descriptors and chain-related descriptors contributed significantly to solubility parameter prediction, and these descriptors were therefore taken into the PolyDrug workflow. Molecular descriptor generation was carried out using PaDEL descriptors.[3]
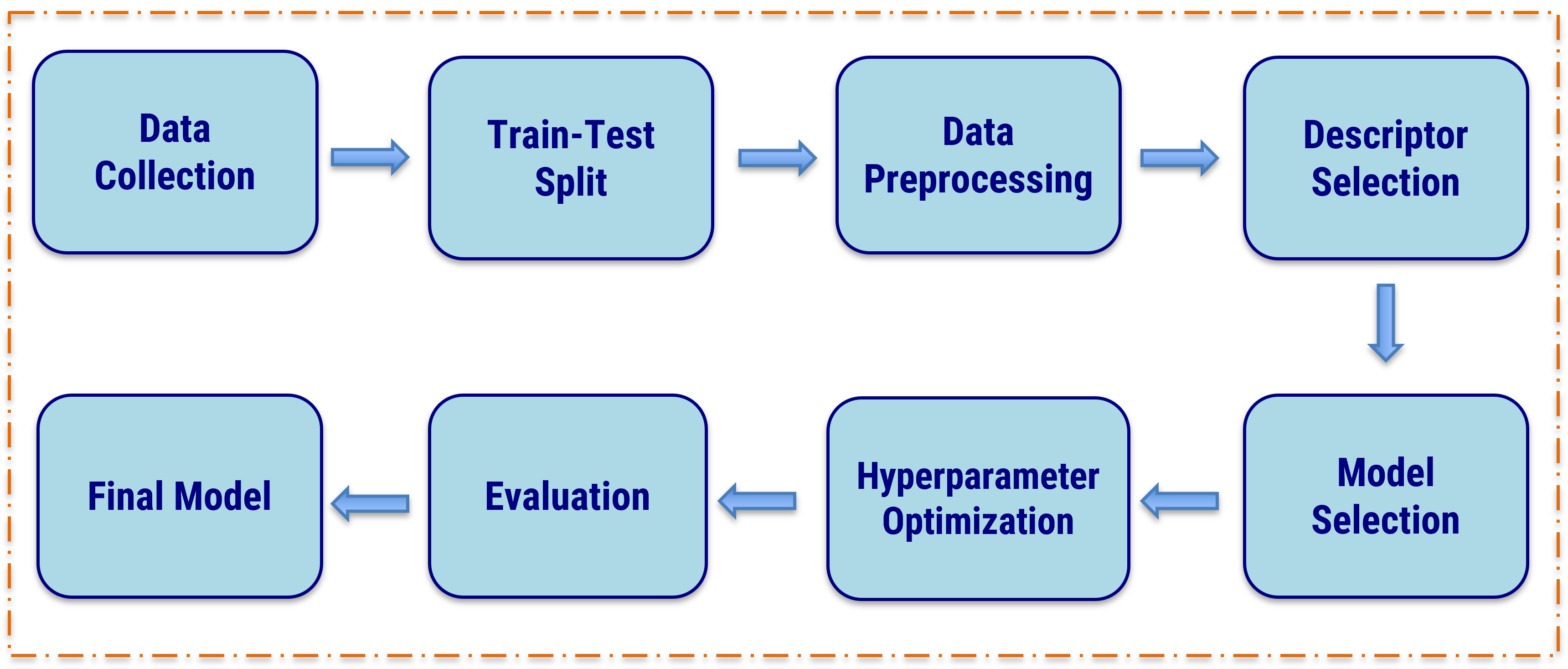
Figure 1: The workflow figure summarizes the main steps used in the study, from molecular representation and descriptor generation to feature selection and predictive modeling.
Pearson Correlation Coefficient (PCC)¶
To identify the most relevant input variables, Pearson correlation coefficient (PCC) analysis was used. The Pearson correlation coefficient measures the strength and direction of the linear relationship between two continuous variables, with values ranging from -1 to +1. A value of +1 indicates a perfect positive linear relationship, -1 indicates a perfect negative linear relationship, and 0 indicates no linear correlation.
The coefficient is defined as:
In this expression, \(r\) is the correlation coefficient, \(x_i\) and \(y_i\) are the observed values, and \(\bar{x}\) and \(\bar{y}\) are the corresponding mean values. In the feature selection workflow, PCC was used to evaluate the linear correlation between each descriptor and the target solubility parameter. Only descriptors showing moderate or strong correlation (PCC > 0.5) were retained, reducing the feature set to 43 descriptors and removing weak or redundant variables. The PCC figure below highlights the descriptors most strongly related to the solubility parameter and provides an informative basis for predictive machine learning models.
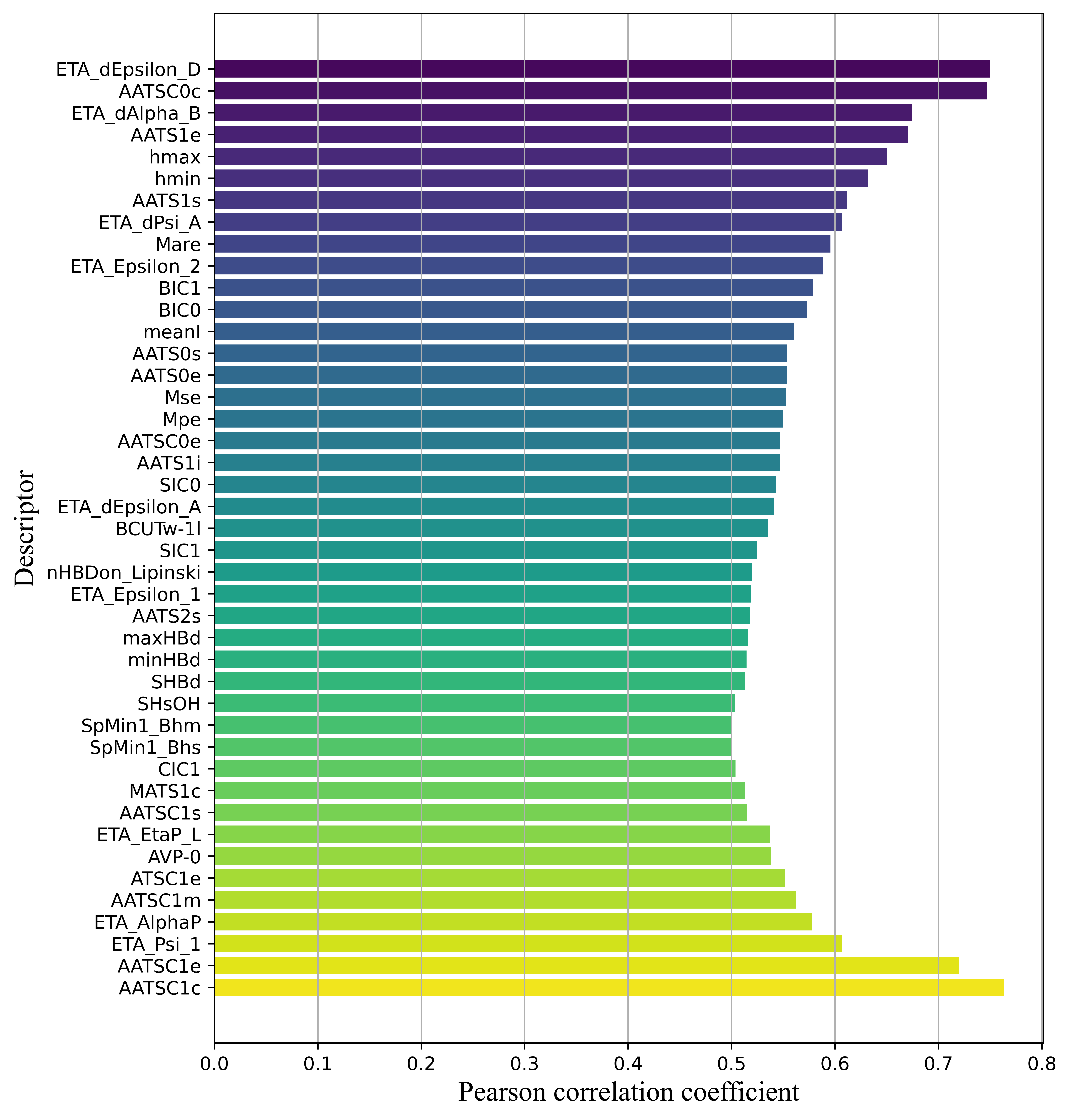
Figure 2: PCC analysis of the descriptors used for solubility parameter prediction, showing the descriptors retained after correlation filtering. Further details of the descriptors are available in Refs. [1,3]
Random Forest Model¶
Random Forest (RF) showed strong and well-balanced performance across the evaluated feature-selection strategies.[1] In particular, the RF model combined with PCC provided reliable results on both training and test data, indicating good predictive accuracy together with stable generalization.
As a tree-based ensemble method, RF is well suited for descriptor-based molecular datasets because it can capture nonlinear relationships between descriptors and the target property while reducing overfitting relative to a single decision tree. This makes RF a practical and robust choice for solubility parameter prediction in PolyDrug.
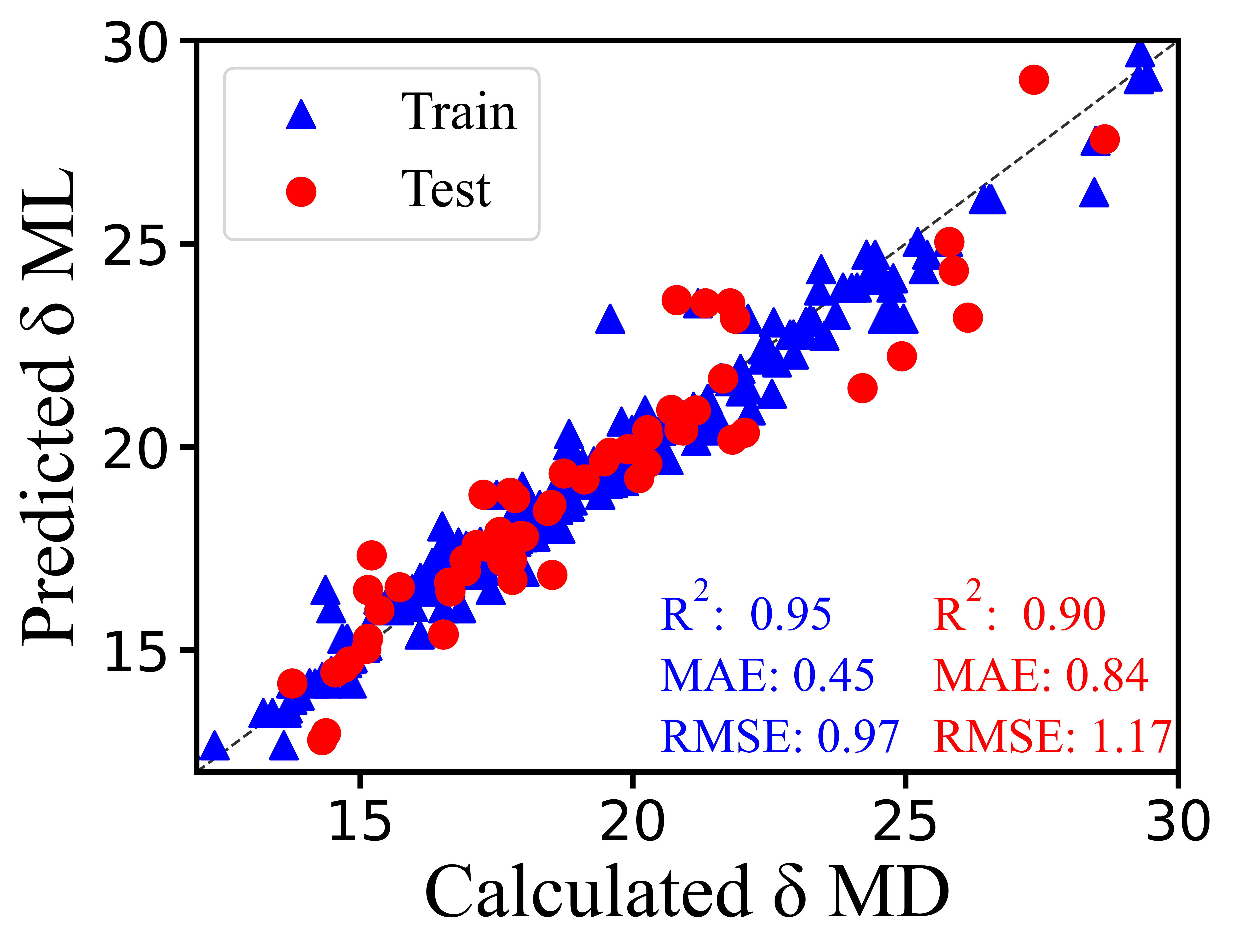
Figure 3: Performance of RF models with PCC feature selection for solubility parameter prediction. MAE and RMSE are reported in MPa½.
References¶
-
George A, Sierka M. Machine Learning Meets Molecular Dynamics: Accurate Prediction of Polymer Solubility Parameters. Advanced Theory and Simulations. 2026;9(3):e01865. https://doi.org/10.1002/adts.202501865
-
Chi M, Gargouri R, Schrader T, Damak K, Maalej R, Sierka M. Atomistic Descriptors for Machine Learning Models of Solubility Parameters for Small Molecules and Polymers. Polymers. 2022;14(1):26. https://doi.org/10.3390/polym14010026
-
Yap CW. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. Journal of Computational Chemistry. 2011;32(7):1466-1474. https://doi.org/10.1002/jcc.21707