MD-ML Hybrid¶
This section summarizes the hybrid molecular dynamics and machine learning strategy used in PolyDrug for solubility parameter prediction.
MD Workflow¶
The solubility parameters for small molecules, polymers and drugs were first determined through a systematic atomistic molecular dynamics (MD) workflow.[1,2,3] Initial amorphous models were generated using an amorphous-cell procedure based on a conformational Monte Carlo protocol. Small-molecule systems contained hundreds of molecules per simulation cell, while polymer systems contained multiple chains depending on polymer chain length. For each system, several initial configurations were generated to ensure adequate conformational sampling, and low-energy structures were selected for subsequent equilibration.
The equilibration stage followed a multi-step workflow. Selected structures first underwent simulated annealing to overcome local energy barriers and identify more stable conformations. This was followed by NVT and NPT equilibration steps and a final sampling stage used to calculate thermodynamic properties. After equilibration, the systems were used to calculate properties such as mass density \(\rho\), solubility parameter \(\delta\), and thermodynamic compatibility.
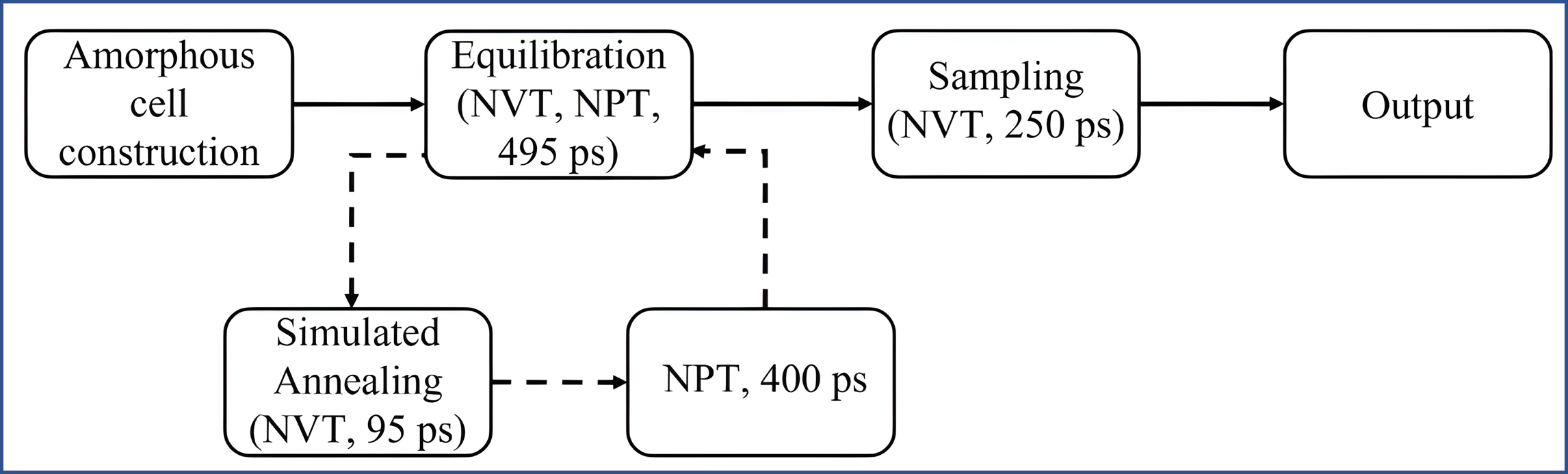
Figure 1: Overview of the MD simulation workflow for solubility parameter calculation, including amorphous-cell construction, equilibration, and sampling steps. [1]
MD-ML Hybrid approach¶
MD simulations can provide reliable reference data, but their computational cost limits their use for rapid large-scale screening. To overcome this limitation, MD-derived solubility parameter data were used as training targets for machine learning models.

Figure 2: Overview of the MD-ML hybrid approach. MD simulations provide mass density \(\rho\), solubility parameter \(\delta\), and thermodynamic compatibility data, which are then used for machine learning model development. [1]
This MD-ML strategy combines the physical detail of atomistic simulations with the speed of machine learning. In PolyDrug, the hybrid approach enables faster prediction of solubility-related properties while retaining a close connection to simulation-based reference data. The MD results provide the physically motivated target values, and the ML models then learn the relationship between descriptor-based inputs and those MD-derived properties.
References¶
-
George A, Sierka M. Machine Learning Meets Molecular Dynamics: Accurate Prediction of Polymer Solubility Parameters. Advanced Theory and Simulations. 2026;9(3):e01865. https://doi.org/10.1002/adts.202501865
-
Westhoff J, Weber C, Bachmann V, Göppert NE, Peric M, George A, Sierka M, Hoeppener S, Vilotijevic I, Schubert US, Werz O, Fischer D. Amino acid-equipped poly(ester amide) nanoparticles for improved encapsulation efficiency of lipophilic anti-inflammatory 6BIGOE based on molecular dynamic simulations. International Journal of Pharmaceutics. 2025;684:126182. https://doi.org/10.1016/j.ijpharm.2025.126182
-
Klepsch LC, Dahlke P, Behnke M, Tsarenko E, Göppert NE, Klemm P, Meyer J, George A, Chi M, Czaplewska JA, Vollrath A, Weber C, Jordan PM, Schubert S, Hoeppener S, Nischang I, Sierka M, Werz O, Schubert US. Matching drug and polymer for efficient delivery of anti-inflammatory drugs: PLGA, polyesterolamides, and acetylated dextran. Journal of Materials Chemistry B. 2025. https://doi.org/10.1039/D5TB01949D